NSAIDsの種類と鎮痛効果の強さについて医学的根拠に基づいて解説します。COX阻害の選択性や剤形による効果の違いも紹介。慢性痛や急性痛に対して最適なNSAIDsはどれでしょうか?
有害事象報告における医療従事者の責務と効果的実施手順
医療現場で発生する有害事象について、医療従事者が適切に報告するための法的義務や手順、安全管理への重要性を詳しく解説します。正確な報告が医療安全にどのような影響をもたらすのでしょうか?
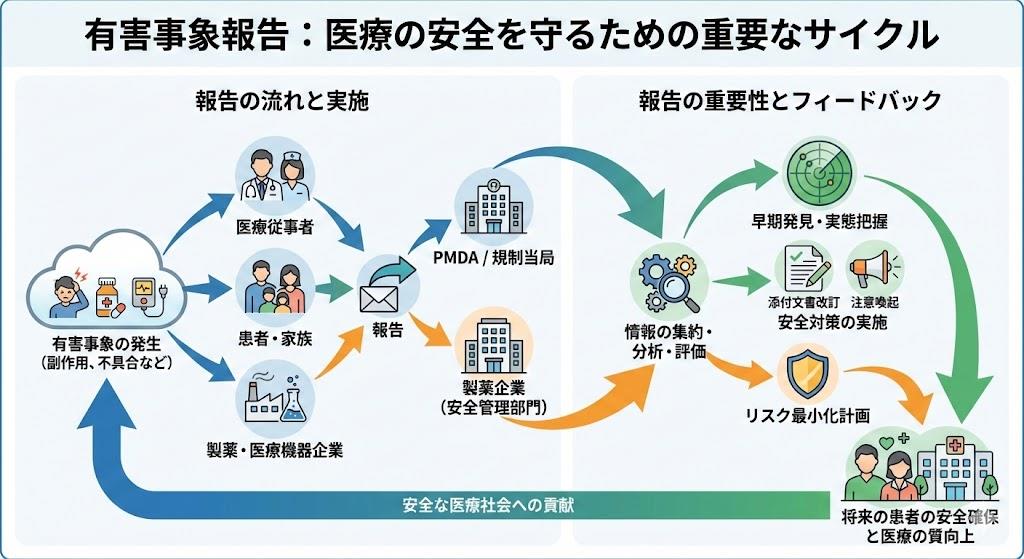
有害事象報告制度は、医療現場における患者安全の確保において不可欠な仕組みです。この制度は医薬品医療機器等法第68条の10第2項に基づき、医療従事者に法的義務として課せられています。医薬品・医療機器の使用によって発生する健康被害等の情報を収集し、医薬品等の安全性確保に活用することが主たる目的です。
すべての医療機関および薬局等を対象とし、薬局開設者、病院若しくは診療所の開設者、または医師、歯科医師、薬剤師、登録販売者その他病院等において医療に携わる者のうち業務上医薬品、医療機器又は再生医療等製品を取り扱う方が報告者となります。
報告対象となる有害事象は以下の通りです。
特に重要なのは、医薬品関連のヒヤリ・ハット報告において約30%を占める医薬品関連事故への対策です。これらの報告により、医薬品安全管理責任者の設置等、医薬品関連の安全確保の制度化が順次進んでいます。

医薬品医療機器等安全性情報報告制度は、医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律に基づく重要な制度です。報告者の範囲は幅広く設定されており、医療に直接携わる職種だけでなく、業務上医薬品や医療機器を取り扱うすべての関係者が対象となります。
この制度の特徴は、報告文化の醸成にあります。医療従事者が患者安全に関する懸念を公平性をもって、非難を恐れることなく報告できる雰囲気の醸成が重要とされています。機密性が保証され、従業員が提出した報告書が改善のために活用され、その報告が価値のある行為として認識される環境作りが求められます。
報告制度の効果として、これまで知られていなかった副作用の発見や、医薬関係者および患者への注意喚起が可能になります。たくさんの副作用報告をいただくことで、より安全に医薬品を使用できるようになると考えられています。
有害事象報告の手順は、発生した事象の重篤度により異なります。重篤な有害事象の場合、特に厳格な報告期限が定められています:
一次報告(72時間以内)
二次報告(7日以内)
報告方法については、複数の選択肢が用意されています:
電子的な報告システムでは、報告書の作成からPMDAへの提出までオンラインで可能となっており、効率的な報告が実現されています。
有害事象報告において最も重要な要素の一つが因果関係評価です。報告者である医療従事者の意見は、企業や規制当局による評価において重要な情報として扱われます。実際に患者を診ている報告者の意見は、客観的なデータと同様に価値のある情報とされています。
因果関係評価において考慮される要素。
医師以外の医療従事者の意見についても、実際に患者ケアに関わる専門職として重要な情報源と位置づけられています。そのため、報告者の判定にばらつきが生じにくい判定基準や選択肢を設けることが重要であるとされています。
有害事象報告データは、医療安全向上のための貴重な情報源として活用されています。米国FDAが運営するFAERS(FDA Adverse Event Reporting System)では、1000万件もの有害事象発症例が世界中から公開されており、誰もがアクセス可能な形で提供されています。
このデータベースの特徴。
日本においても、患者さんからの直接的な副作用報告制度が導入されています。これは「薬害肝炎事件の検証及び再発防止のための医薬品行政のあり方検討委員会」の最終提言を受けて実施されているもので、多様な観点からの報告を活用することの有用性が認識されています。
効果的な有害事象報告には、個人レベルの対応だけでなく、医療機関全体での組織的な取り組みが不可欠です。当院における有害事象報告体制では、以下のような手順が確立されています:
組織内報告体制の構築
報告書式の標準化
重篤な有害事象の定義も明確に定められており、死亡、死亡につながるおそれのある症例、入院又は入院期間の延長が必要とされる症例、障害、障害につながるおそれのある症例などが含まれます。これらの定義により、報告すべき事象の判断基準が統一されています。
がん化学療法領域では、特に重篤有害事象報告体制の構築が進んでいます。化学療法に関連した重篤有害事象の発現状況を詳細に把握し、適切な対応策を講じるための専門的な報告システムが確立されています。
有害事象報告制度は、単なる法的義務を超えて、医療の質向上と患者安全確保のための重要な仕組みです。医療従事者一人ひとりが制度の意義を理解し、適切な報告を行うことで、より安全で効果的な医療の提供が可能になります。継続的な報告と分析により、未知の副作用の早期発見や安全対策の構築が実現され、最終的には患者さんの安全と医療の質向上に貢献することができるのです。
PMDAの医薬品医療機器等法に関する報告制度について - 報告制度の詳細と手続きに関する包括的な情報
厚生労働省の医薬品副作用・医療機器不具合報告ガイドライン - 報告対象となる事象の具体的な判断基準
[指定医薬部外品] 大正製薬 新ビオフェルミンS錠 550錠 61日分整腸剤【Amazon.co.jp限定】 [乳酸菌/ビフィズス菌/フェーカリス菌/アシドフィルス菌 配合] 腸内フローラ改善 便秘や軟便に